
Diagnóstico genético de enfermedades gastrointestinales
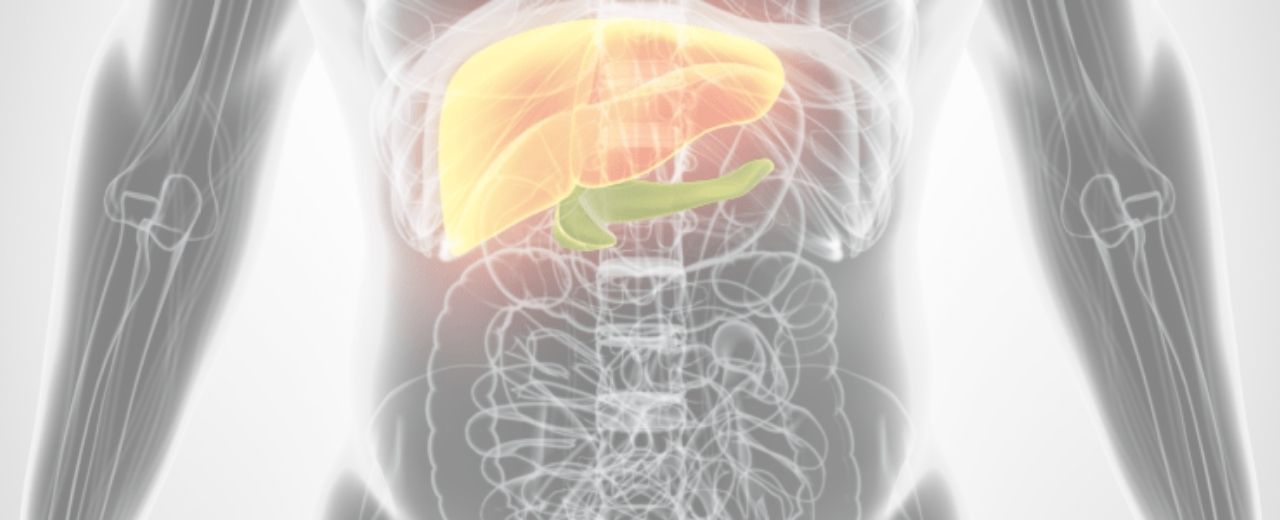
Hígado
En el recién nacido o lactante, las causas de hepatopatía predominantes son: la inflamación idiopática (atresia biliar) y los trastornos genéticos (colestasis intrahepática, déficit de alfa-1-antitripsina, metabolopatías, etc.); y la presentación habitual la de un cuadro de ictericia colestática que, en un porcentaje pequeño de casos, asocia una situación de fallo hepático. En el lactante (mayor de 6 meses) o niño mayor, las posibles causas de hepatopatía son más variadas (congénitas o adquiridas) y se detectan frecuentemente de forma casual o ante síntomas inespecíficos (astenia, anorexia, dolor abdominal, etc.). Es en este grupo de niños donde, con frecuencia, se detecta una elevación de transaminasas y, con menos frecuencia, una ictericia colestática, lo que constituye el punto de partida para el diagnóstico de su enfermedad hepática. Otras posibles causas genéticas de ictericia son: defecto en la síntesis de ácidos biliares colestasis neonatal colelitiasis colangitis esclerosante.
La deficiencia de alfa-1-antitripsina afecta a 1/2.000 recién nacidos vivos, de los cuales, un 10-20% desarrollarán una hepatopatía de grado variable durante la infancia. Una mutación en el gen SERPINA1 condiciona la producción de una alfa-1-antitripsina anómala que se acumula en las células dañándolas.
Diferentes enfermedades metabólicas pueden producir una ictericia-colestasis de inicio precoz; si bien, esta no suele ser el síntoma predominante, sino otros hallazgos, como fallo o insuficiencia hepática, con déficit de la actividad de protrombina, que no se corrige tras administración de vitamina K, hipoglucemia y colinesterasa baja. Es el caso de la galactosemia tirosinemia o fructosemia.
Las enfermedades de almacenamiento de glucógeno (glucogenosis), o lípidos (enfermedad de Nieman-Pick), y enfermedad de Wolman (déficit de lipasa ácida) producen hepatomegalia.
Otras enfermedades hepáticas son causadas por almacenamiento de metales, cuyo metabolismo normal está alterado. La hemocromatosis es una enfermedad en la que el hierro se acumula en el hígado lo que provoca enfermedad hepática. En la enfermedad de Wilson el metal que se acumula es el cobre. En ambos casos es de extrema importancia para el pronóstico de la enfermedad un diagnóstico temprano.
Algunos tipos de defectos en la glicosilación de proteínas, algunas enfermedades mitocondriales, defectos de oxidación de ácidos grasos, trastornos peroxisomales y los trastornos del ciclo de la urea presentan también graves complicaciones gastrointestinales.
La enfermedad poliquística del hígado es el resultado de una malformación de la placa ductal embrionaria del árbol biliar intrahepático. El fenotipo consta de numerosos quistes diseminados por el parénquima hepático. El cribado de mutaciones de los genes que la causan confirman el diagnóstico clínico.
Páncreas
La fibrosis quística es un trastorno autosómico recesivo que limita la vida y que afecta a 70.000 personas en todo el mundo principalmente en poblaciones de ascendencia europea. Causa daños graves en los pulmones, el sistema digestivo y otros órganos del cuerpo. La fibrosis quística afecta a las células que producen moco, sudor y jugos digestivos. Estos líquidos secretados son normalmente ligeros y resbaladizos. Pero en las personas con fibrosis quística, un gen defectuoso hace que las secreciones se vuelvan pegajosas y espesas. En lugar de actuar como lubricantes, las secreciones tapan los tubos, conductos y pasajes, especialmente en los pulmones y el páncreas.
Además, existen otras enfermedades genéticas que pueden afectar al páncreas: pancreatisis hereditarias deficiencia de lipasa pancreática etc.
Intestino
Los trastornos diarreicos congénitos (CDD) representan una red en evolución de enteropatías crónicas raras, con un inicio típico temprano en la vida. En muchas de estas afecciones, la diarrea crónica grave representa la manifestación clínica primaria, mientras que en otras la diarrea es solo un componente de un trastorno sistémico o multiorgánico más complejo. Por lo general, durante los primeros días de vida, la diarrea conduce a una afección potencialmente mortal resaltada por una deshidratación grave y anomalías de los electrolitos séricos. Por lo tanto, en la gran mayoría de los casos, se debe iniciar de inmediato la terapia adecuada para prevenir la deshidratación y las complicaciones a largo plazo, a veces graves. El número de trastornos genéticos bien caracterizados atribuidos a estos trastornos ha aumentado gradualmente en los últimos años, se han identificado muchos genes nuevos y se han relacionado funcionalmente con las CDD, lo que abre nuevas perspectivas diagnósticas y terapéuticas.
El análisis molecular ha cambiado el escenario de diagnóstico en las CDD y ha llevado a una reducción de los procedimientos invasivos y costosos. Se han logrado importantes avances en términos de patogenia, lo que permite una mejor comprensión no solo de estas condiciones raras sino también de los mecanismos de enfermedades más comunes.
Hay pacientes con un espectro diverso de trastornos genéticos raros que pueden presentar enfermedades inflamatorias del intestino. Los pacientes con estos trastornos a menudo desarrollan síntomas durante la infancia o la niñez temprana, junto con características endoscópicas o histológicas de la enfermedad de Crohn, colitis ulcerosa o enfermedad inflamatoria intestinal sin clasificar. Los defectos en la señalización de la interleucina 10 tienen un patrón de herencia mendeliano con una penetración completa de la inflamación intestinal. Varios defectos genéticos que alteran la función de la barrera epitelial intestinal o afectan la función inmune innata y adaptativa tienen una penetrancia incompleta del fenotipo similar a la enfermedad inflamatoria intestinal. Varias de estas condiciones monogénicas no responden a la terapia convencional y están asociadas con una alta morbilidad y mortalidad.
Debido al amplio espectro de estas enfermedades extremadamente raras, un diagnóstico correcto suele ser un desafío y, a menudo, llegar con retraso. En muchos casos, estas enfermedades no se pueden clasificar según las características histológicas e inmunológicas estándar de la enfermedad inflamatoria intestinal. Se requiere un análisis genético para identificar la causa del trastorno y ofrecer al paciente opciones de tratamiento apropiadas, que incluyen terapia médica, cirugía o trasplante alogénico de células madre hematopoyéticas. Además, el diagnóstico basado en el análisis genético puede conducir al asesoramiento genético para los familiares de los pacientes.
Otros síndromes con afectación gastrointestinal
Por último, numerosos síndromes multisistémicos presentan afectación gastrointestinal importante: Dubin-Johnson, Shwachman-Diamond, SIDDIQI, Rajab, Alagille, FINCA, Omenn, Mitchell-Riley, síndrome del intestino corto etc…
Consulta nuestras pruebas genéticas para la ayuda al diagnóstico genético de enfermedades del sistema gastrointestinal:
¿Necesitas preguntarnos por estas pruebas? No dudes en ponerte en contacto.
Correo electrónico: info@genome4care.com